Indikation
In Deutschland sind zwei Natalizumab-haltige Präparate (TYSABRI® und TYRUKO®) zugelassen.
Natalizumab ist in Deutschland zur krankheitsmodifizierenden Monotherapie der aktiven, schubförmig verlaufenden Multiplen Sklerose zugelassen.
Natalizumab kann bei den folgenden Patientengruppen eingesetzt werden:
Erwachsene Patienten ab 18 Jahren, die trotz Behandlung mit einem vollständigen und angemessenen Zyklus mit mindestens einer krankheitsmodifizierenden Therapie (DMT) eine fortgesetzte Krankheitsaktivität aufweisen.
Erwachsene Patienten ab 18 Jahren mit rasch fortschreitender schwerer schubförmig-remittierend verlaufender Multipler Sklerose können auch primär mit Natalizumab behandelt werden, wenn…
| … zwei oder mehr Schübe mit Behinderungsprogression in einem Jahr aufgetreten sind und |
| … in der kranialen MRT mindestens eine neue Gadolinium-anreichernde Läsion oder eine relevante Zunahme der T2-Läsionen im Vergleich zu einer kürzlich durchgeführten MRT-Aufnahme nachweisbar sind. |
Kontraindikationen
Natalizumab ist kontraindiziert bei …
- Überempfindlichkeit gegen die Substanz oder einen der sonstigen Bestandteile (beschrieben wurden ca. 4% leichte bzw. 0,8% schwere Hypersensitivitäts-Reaktionen).
- Patienten mit progressiver multifokaler Leukenzephalopathie (PML, aktuell oder in der Vorgeschichte).
- Patienten mit einem erhöhten Risiko für opportunistische Infektionen, wie immungeschwächte Patienten (einschließlich solcher Patienten, die aktuell eine immunsuppressive Behandlung erhalten oder durch frühere Therapien immungeschwächt sind).
- vorliegender HIV-Infektion.
- aktiven Malignomen mit Ausnahme von Patienten mit einem Basaliom.
- Patienten, die das 18. Lebensjahr noch nicht vollendet haben. Die Behandlung sollte nur nach kritischer Nutzen-Risiko-Bewertung erfolgen. Die Therapie sollte durch ein erfahrenes Zentrum durchgeführt werden.
Verfügbare Präparate
Im Juni 2006 wurde in der Europäischen Union erstmals das Natalizumab-haltige Präparat TYSABRI® zur intravenösen Infusion zugelassen.
Im März 2021 wurde TYSABRI® zur subkutanen Anwendung in der Europäischen Union zugelassen.
Im September 2023 wurde mit TYRUKO® das erste Natalizumab-haltige Biosimilar zur intravenösen Infusion zugelassen. Die Zulassung von TYRUKO® basiert neben den frühen klinischen und präklinischen Daten insbesondere auf dem Nachweis gleichwertiger Wirksamkeit gegenüber TYSABRI® in der ANTELOPE-Studie (doi: 10.1001/jamaneurol.2022.5007). Zwischen TYRUKO® und TYSABRI® bestehen formal kleinere Unterschiede in der Herstellung (TYSABRI®: Herstellung in Maus-Zellen; TYRUKO®: Herstellung in Hamster-Zellen); dies hat nach aktuellem Kenntnisstand keine praktischen Konsequenzen.
Die Fachinformationen zwischen TYSABRI® und TYRUKO® gleichen sich in den Abschnitten „Darreichungsform“, „Klinische Angaben“ und „Pharmakologische Eigenschaften“ im Wortlaut.
Dennoch existieren einerseits regulatorische Unterschiede zwischen den Präparaten bezüglich Schulungsmaterialien und Patientenaufklärungen.
Dosierung
TYRUKO®/TYSABRI®
Natalizumab wird als Infusion zu 300 mg alle vier Wochen intravenös verabreicht. Jede Durchstechflasche enthält 15ml Konzentrat (20 mg/ml) und wird vor Applikation mit 100 ml 0,9%iger NaCl-Lösung verdünnt.
Dosisanpassungen nach Gewicht, Geschlecht oder ethnischer Zugehörigkeit müssen nicht vorgenommen werden.
Die subkutane Injektion erfolgt in einer Dosierung von 300 mg (auf 2 Injektionsstellen verteilt) alle 4 Wochen, wobei die Injektionsstellen mindestens 3cm voneinander entfernt liegen sollen.
Ergänzende Informationen (formal zu TYSABRI® erhoben):
Nachdem langjährig erhobene Daten aus dem TOUCH-Register bereits darauf hinwiesen, dass eine Verlängerung des Infusionsintervalls von 4 auf 6 Wochen zu einer Verminderung des PML-Risikos bei erhaltener Wirksamkeit führen könnte (Ryerson LZ, Foley J, Chang I, et al. Reduced Risk of Progressive Multifocal Leukoencephalopathy (PML) Associated with Natalizumab Extended Interval Dosing (EID): Updated Analysis of the TOUCH® Prescribing Program Database. Presented at the American Academy of Neurology 71st Annual Meeting; 4 – 10 May 2019; Philadelphia, PA.), wurden im Juli 2022 die Ergebnisse der NOVA-Studie veröffentlicht (Foley JF et al. Lancet Neurol. 2022 Jul;21(7):608-619. doi: 10.1016/S1474-4422(22)00143-0.). Hier zeigten sich bezüglich der Krankheitsaktivität keine eindeutig über die bei Baseline bereits bestehenden Unterschiede hinausgehenden Effekte. Eine Aussage bezüglich des PML-Risikos konnte in der Studie nicht gemacht werden aufgrund der zu geringen Anzahl an Patientenjahren. Die Entscheidung, das Dosisintervall auf 6 Wochen auszudehnen (Extended Intervall Dosing), sollte weiterhin erfahrenen Zentren vorbehalten sein. Es handelt sich weiterhin um eine off label-Behandlung. Diese Daten zu verlängertem Dosisintervall beziehen sich ausschließlich auf die intravenöse Gabe von TYSABRI®.
Pharmakokinetik
- Nach wiederholter Gabe von 300 mg Natalizumab werden maximale Serumkonzentrationen von ca. 110 ± 52μg/ml erreicht. Der durchschnittliche Talspiegel im steady state liegt bei 23 – 29μg/ml. Die mittlere Plasmahalbwertszeit liegt bei 16 ± 4 Tagen.
- Das Auftreten persistierender neutralisierender Antikörper erhöht die Ausscheidung des Medikaments um das Dreifache (Auftreten in ca. 6% der Patienten in den ersten drei Monaten, selten später als nach neun Monaten).
- Mittels Plasmaaustausch oder Immunadsorption kann Natalizumab schneller aus dem Organismus eliminiert werden.
Pharmakodynamik
- Natalizumab bindet nach Infusion an das Adhäsionsmolekül α4-Integrin, welches auf einer Vielzahl von Immunzellen, insbesondere jedoch auf T- und B-Lymphozyten, exprimiert wird.
- Als IgG4-Antikörper löst Natalizumab keinen Zelltod aus, verhindert aber die Adhäsion von Immunzellen an der Gefäßwand und dadurch schlussendlich die Überwindung der Blut-Hirn-Schranke.
- Die Wirkung von Natalizumab auf das Immunsystem ist weder organspezifisch (so wird auch die Einwanderung von Immunzellen in andere Organe wie den Darm gehemmt) noch selektiv für autoreaktive Immunzellen. Dementsprechend wird auch die Einwanderung von Immunzellen, welche spezifische Krankheitserreger bekämpfen, behindert.
- Aufgrund des Wirkmechanismus zeigen Patienten unter Therapie mit Natalizumab tendenziell höhere Leukozytenzahlen im peripheren Blut. Nach Absetzen des Medikaments und Freigabe des α4-Integrins können diese Zellen rasch in das ZNS einwandern und ggfs. wiederkehrende Krankheitsaktivität bedingen.
Diagnostik || vor Therapiebeginn
Anamnese und klinische Untersuchung zu möglichen Kontraindikationen
Durch eine sorgfältige Anamnese und klinische Untersuchung sollte gezielt nach dem Vorliegen möglicher Kontraindikationen gesucht werden. Anamnese und Untersuchung müssen detailliert dokumentiert werden (obligat).
Labor-Basisprogramm
- Routinelaborparameter: Die Bestimmung von Blutbild plus Differenzialblutbild und Leberwerten (GOT, GPT, GGT) ist obligat.
- Entzündungs- und Infektionsparameter: Vor Beginn der Therapie mit Natalizumab sollten akute Infektionen (CRP fakultativ) und chronische aktive bakterielle und virale Infektionen ausgeschlossen werden. Eine Hepatitis-B- und -C-Serologie sowie VZV-Serologie können erhoben werden (fakultativ). Bei Verdacht auf Tuberkulose (Tbc) in der Vorgeschichte oder bei Personen mit erhöhter individueller Risikosituation sollte ein Tbc-Test erfolgen (vorzugsweise als IGRA-Test, z.B. Quantiferon(R) oder T-Spot.TB(R)) (obligat). Bei positivem Testergebnis muss die Gefahr einer Tbc-Reaktivierung abgeklärt werden (Röntgen-Thorax und ggf. weitere Diagnostik) (obligat). Ein HIV-Test ist obligat. Zur Durchführung der HIV-Serologie ist eine Einverständniserklärung der Patienten erforderlich.
- Schwangerschaftstest: Bei Patientinnen wird ein Schwangerschaftstest empfohlen (fakultativ).
Radiologische Diagnostik
Eine Referenz-MRT des Schädels vor und nach Gabe von gadoliniumhaltigem Kontrastmittel muss vor Behandlungsbeginn mit Natalizumab angefertigt werden (nicht älter als drei Monate) mit dem Ziel, im Falle einer Krankheitsprogression oder im Falle unvorhergesehener Nebenwirkungen über einen Ausgangsbefund zu verfügen (obligat).
Dokumentierte Aufklärung der Patienten über Therapie und Risiken
Eine standardisierte Aufklärung mit schriftlicher Einwilligungserklärung zur Therapie muss vorliegen (obligat). Über mögliche spezifische Nebenwirkungen und entsprechende Vorsichtsmaßnahmen muss aufgeklärt werden. Hierbei muss insbesondere auf folgende Aspekte eingegangen werden:
- Zu den häufigsten Nebenwirkungen zählen Kopfschmerzen, Harnwegsinfektionen, Atemwegserkrankungen, evtl. in Verbindung mit Schmerzen. Auch vermehrtes Auftreten von Völlegefühl wurde beschrieben. Leichter Schwindel und Übelkeit können während oder kurz nach der Infusion / Injektion auftreten.
- In seltenen Fällen wurden allergische Reaktionen beobachtet, das Gleiche gilt für schwere Infektionen, die ebenfalls selten unter Natalizumab auftreten können.
- Weiterhin wurden Fälle einer Thrombozytopenie in Zusammenhang mit Natalizumab berichtet. Die Diagnose einer solchen kann mit schweren Folgen einhergehen, so dass im Falle einer Thrombozytopenie ein Absetzen erwogen werden kann.
- Mit Stand August 2023 wurden 919 Fälle einer PML (Progressive multifokale Leukenzephalopathie) nach Einnahme von Natalizumab gemeldet. Die PML ist bisher nicht spezifisch behandelbar und kann zu schwerer Behinderung oder sogar zum Tod führen. Das PML-Risiko beträgt derzeit 3,48:1.000. Das individuelle Risiko hängt jedoch deutlich von der Gesamtsituation des Patienten ab.
- Neben der dokumentierten Aufklärung vor der Therapie, ist unter Therapie mit Natalizumab auch die explizite Aufklärung über die Langzeit-Risiken (d.h. bei Behandlung jenseits des 24. Monats) erforderlich und empfohlen. Hierfür stellt das KKNMS einen zusätzlichen Patientenaufklärungsbogen bereit (s. Ende des Kapitels).
- Bezüglich der Therapie mit Natalizumab enthalten die behördlichen Unterlagen des Paul-Ehrlich-Instituts auch ein Aufklärungsformular zum Absetzen der Therapie. Hierfür stellt das KKNMS einen weiteren Patientenaufklärungsbogen bereit (s. Ende des Kapitels).
- Die schriftliche Aufklärung und Dokumentation wird aus formalen Gründen aktuell auch bei Umstellung von TYSABRI® auf TYRUKO® bzw. vice versa empfohlen.
- Die Aushändigung eines Patientenpasses soll bei jedem Patienten, der ein Natalizumab-haltiges Präparat erhält, erfolgen.
Vortherapien || Abstand und Maßnahmen
Behandlungsnaive Patienten
Es ist keine weitere Zusatzdiagnostik über die o.g. Maßnahmen hinaus nötig.
Vorbehandlung mit Glatirameracetat oder Interferon-beta-Präparaten
Ein Sicherheitsabstand ist nicht notwendig, die Behandlung kann direkt erfolgen, vorausgesetzt, eventuelle Effekte jener Therapien auf das Immunsystem (z.B. Zytopenie) sind abgeklungen. Keine weitere Zusatzdiagnostik notwendig.
Vorbehandlung mit Dimethylfumarat/Diroximelfumarat
Ein Sicherheitsabstand ist nicht notwendig, die Behandlung kann direkt erfolgen (vorausgesetzt, eventuelle Effekte der Therapie auf das Immunsystem (z.B. Zytopenie) sind abgeklungen (obligat). Die periphere Immunkompetenz muss bei den in Frage kommenden Patienten hergestellt sein, soweit dies im Differenzialblutbild nachweisbar ist (obligat).
Vorbehandlung mit Teriflunomid
Es wird empfohlen, eine aktive Auswaschung vorzunehmen. Andernfalls muss ein Sicherheitsabstand von mindestens 3,5 Monaten eingehalten werden. Die Autoren empfehlen jedoch grundsätzlich die aktive Auswaschung von Teriflunomid vor Therapiewechsel. Eventuelle Effekte der Therapie auf das Immunsystem (z.B. Zytopenie) müssen abgeklungen sein (obligat). Die periphere Immunkompetenz muss bei den in Frage kommenden Patienten hergestellt sein, soweit dies im Differenzialblutbild nachweisbar ist (obligat).
Vorbehandlung mit Fingolimod, Siponimod, Ozanimod oder Ponesimod
Bei Umstellung von S1P-Rezeptor-Modulatoren auf Natalizumab muss vor Beginn der Therapie ein Sicherheitsabstand eingehalten werden, der sich nach der Eliminationshalbwertszeit der einzelnen Substanzen (bzw. ihrer bioaktiven Metaboliten) bemisst. Ein Sicherheitsabstand von mindestens vier Wochen nach Absetzen des S1P-Rezeptor-Modulators wird für Fingolimod und Ozanimod empfohlen, während dieser Abstand bei Siponimod und Ponesimod kürzer sein kann (ein bis zwei Wochen). Pharmakodynamische Wirkungen (Senkung der Lymphozyten im peripheren Blut) können aber nachhinken. Daher ist zum Ausschluss einer relevanten Lymphopenie ein Blutbild plus Differenzialblutbild durchzuführen (obligat).
Beim Absetzen von Fingolimod ist zu beachten, dass es bei ca. 10% der mit Fingolimod behandelten Patienten zu einem Rebound-Phänomen mit zum Teil fulminant verlaufenden Schüben kommen kann. In der Regel tritt das Rebound-Phänomen zwei bis vier Monate nach Absetzen von Fingolimod auf. Patienten mit hochaktiver Verlaufsform ihrer MS vor Beginn mit Fingolimod, aber auch Patienten mit unzureichendem Ansprechen auf Fingolimod scheinen eher zu einem Rebound zu neigen. Auch bei anderen S1P-Rezeptor-Modulatoren ist ein Rebound-Phänomen nicht auszuschließen, dabei muss die unterschiedliche Pharmakodynamik der Präparate berücksichtigt werden.
Vorbehandlung mit Azathioprin, Methotrexat oder Mitoxantron
Es wird empfohlen, einen Sicherheitsabstand von mindestens drei Monaten einzuhalten. Ein Differenzialblutbild muss vor Behandlungsbeginn mit Natalizumab erstellt werden (obligat). Eventuelle Effekte jener Therapien auf das Immunsystem (z.B. Zytopenie) müssen abgeklungen sein.
Vorbehandlung mit Cladribin
Wenn aufgrund von Nebenwirkungen oder nicht ausreichender Wirksamkeit der Cladribin-Behandlung auf eine andere Immuntherapie umgestellt wird, ist ein Sicherheitsabstand von mindestens sechs Monaten nach dem letzten Behandlungszyklus einzuhalten. Vor Beginn einer anderen Immuntherapie muss ein Differenzialblutbild einschließlich einer Lymphozytentypisierung (CD4+-T-Zellen, CD8+-T-Zellen, B-Zellen und NK-Zellen) erstellt werden (obligat). Regelmäßige Blutbildkontrollen sollten auch nach Therapieende über mindestens fünf Jahre erfolgen (fakultativ). Therapiespezifische Effekte auf das Immunsystem (z.B. Zytopenie) sollten abgeklungen sein.
Bei Umstellung aufgrund von hoher Krankheitsaktivität unter Cladribin-Behandlung muss im Einzelfall über die Wartezeit entschieden werden, und es sollte eine zeitnahe Vorstellung an einem für MS spezialisierten Zentrum erfolgen.
Vorbehandlung mit Rituximab, Ocrelizumab, Ofatumumab, Alemtuzumab, Ublituximab oder den Immunsuppressiva Ciclosporin A oder Cyclophosphamid
Hier sollte der Sicherheitsabstand vor Beginn der Therapie mit Natalizumab mindestens sechs bis zwölf Monate betragen. Ergänzend muss ein Differenzialblutbild (obligat) erstellt werden, eine durchflusszytometrische Zellphänotypisierung (CD4+-T-Zellen, CD8+-T-Zellen, B-Zellen, NK-Zellen) kann durchgeführt werden (fakultativ). Effekte jener Therapien auf das Immunsystem (z.B. Zytopenie) müssen abgeklungen sein.
Vorbehandlung mit Studienmedikamenten
Hier kann kein spezifischer Sicherheitsabstand festgelegt werden. Effekte jener Therapien auf das Immunsystem (z.B. Zytopenie) sollten abgeklungen sein.
Durchführung || Während der Infusion / Injektion
Die 15 ml des Natalizumab-Konzentrats müssen vor Gebrauch in 100 ml 0,9%iger Kochsalzlösung verdünnt werden und sollen nicht mit anderen Medikamenten in der gleichen Infusion gemischt werden.
Die Infusion sollte in nicht weniger als einer Stunde gegeben werden (obligat), empfohlen ist eine Infusionsgeschwindigkeit von ca. 2 ml/Minute unter Kontrolle der sicheren intravenösen Lage des Katheters während der Infusion. Nach Infusion ist die Infusionsleitung mit 0,9%iger Kochsalzlösung zu spülen.
Paravasate mit der Möglichkeit schwerer lokaler Reaktionen bis hin zu Gewebenekrosen müssen in jedem Fall vermieden werden. Die Infusion muss streng intravenös gegeben werden, d.h. die sichere intravenöse Lage der Venenverweilkanüle muss überprüft werden und die Infusion in eine Hauptvene ist zu bevorzugen. Daneben sollte die Infusion in Extremitäten mit beeinträchtigtem venösem oder lymphatischem Abfluss vermieden werden.
Maßnahmen und Infrastruktur für die Akuttherapie von Paravasaten sollten vorgehalten werden (fakultativ). Bei Auftreten eines Paravasates muss die Infusion sofort unterbrochen werden. Die Kanüle sollte zunächst in situ verbleiben, um hierüber das Paravasat zu aspirieren. Bei Hautblasen oder großem Paravasat sollte dieses transkutan abpunktiert werden. Anschließend sollte DMSO (99%, Dimethylsulfoxid) auf dem gesamten Paravasatgebiet aufgetragen werden. Bei progredienter Gewebenekrose sollte der Patient frühzeitig chirurgisch vorgestellt werden. Nach einem Paravasat sollte der Patient über mindestens sechs Wochen ärztlich nachverfolgt werden.
Bei Auftreten einer Infusionsreaktion muss die Geschwindigkeit reduziert bzw. bei schweren Reaktionen gestoppt werden.
Patienten müssen bis zu einer Stunde nach Beendigung der Infusion weiterhin überwacht werden (obligat).
Mittel zur Behandlung anaphylaktischer und / oder schwerer Reaktionen müssen verfügbar und das Infusionsteam hinsichtlich der Behandlung von anaphylaktischen und / oder schweren Infusionsreaktionen geschult sein (obligat). Ein uneingeschränkter Zugang zu einer intensivmedizinischen Versorgungs- und Behandlungseinheit im eigenen Haus oder im nächstgelegenen Krankenhaus (z.B. via Rettungstransportwagen) ist nach der Erstversorgung einer schweren Infusions- oder allergischen Reaktion erforderlich (obligat).
Häufig tritt während oder innerhalb von einer Stunde nach der Behandlung mit Natalizumab ein Schwindelgefühl auf. Patienten sollte für die Dauer dieser Beschwerden von der aktiven Teilnahme am Straßenverkehr und vom Führen von Maschinen abgeraten werden.
TYSABRI® subkutan:
Die subkutane Injektion von Natalizumab erfolgt durch 2 Injektionen durch Fertigspritzen à 150 mg/ml an 2 verschiedenen Injektionsstellen alle 4 Wochen durch eine medizinische Fachkraft. Die Patienten müssen eine Stunde nach den Injektionen nachbeobachtet werden, nach sechs Gaben kann die Nachbeobachtung nach klinischem Ermessen erfolgen. Die Injektionen sollten möglichst unmittelbar nacheinander erfolgen, die zweite Injektion sollte nicht später als 30 Minuten nach der ersten Injektion gegeben werden. Geeignete Injektionsstellen sind die Rückseite des Oberarms, der Bauch und die Oberschenkel. Die Haut der Injektionsstelle sollte nicht gerötet, verletzt, gereizt oder vernarbt sein. Die Injektionsstellen sollten mehr als 3 cm voneinander entfernt sein.
Seit Januar 2024 besteht bezüglich der subkutanen Therapie mit TYSABRI® die Möglichkeit, bei Patienten, die die letzten 6 vorangegangenen Gaben gut vertragen haben (d.h. keine Infusionsreaktionen zeigten), eine Therapie außerhalb der Praxis/Klinik anzubieten („outside clinical setting“ (OCS)-Therapie). Diese Entscheidung obliegt dem behandelnden Facharzt, dieser entscheidet und verantwortet auch die Delegation an jeweiliges medizinisches Fachpersonal.
Auf der Website des Paul-Ehrlich-Instituts stehen die erforderlichen Fragebögen/Checklisten zur Verfügung, die vor jeder OCS-Injektion abzuarbeiten sind.
Eine Selbstinjektion durch den Patienten ist weiterhin nicht vorgesehen, da die Behandlung weiterhin durch medizinisches Fachpersonal zu erfolgen hat.
Monitoring & Maßnahmen || unter Therapie
Klinisch-neurologische Kontrolle
Vierteljährlich müssen klinisch-neurologische Kontrolluntersuchungen durchgeführt werden (obligat).
Labor-Basisprogramm
- Routinelaborparameter: Ein Blutbild muss regelmäßig bestimmt werden (alle drei bis sechs Monate) (obligat). Vor jeder Natalizumab-Infusion / -Injektion muss klinisch eine Infektkonstellation ausgeschlossen werden. Im Falle einer Leukopenie oder einer deutlichen Leukozytose ist ein Differenzialblutbild notwendig (obligat).
Die Behandlung mit Natalizumab führt zu Blutbildveränderungen. Aufgrund des Wirkmechanismus kann es insbesondere zu einem Anstieg der peripheren Leukozyten kommen. Die Frequenz der CD34+-Stammzellen sowie B-Zellen in der Peripherie steigt an. Zudem kann es zu einer Linksverschiebung im Blutbild kommen. Sehr selten kommt es zu einem deutlichen Abfall von CD4+-Zellen, die dann als Zeichen einer Beeinträchtigung der Immunkompetenz zu werten wären. In diesen Fällen sollte Rücksprache mit einem qualifizierten Zentrum erfolgen und die Therapie mit Natalizumab zunächst nicht fortgeführt werden.
- Eine einmalige Bestimmung der Leberwerte (GOT, GPT, GGT) innerhalb von drei Monaten nach Therapiestart ist obligat. Danach sollten sie alle drei Monate im ersten halben Jahr unter laufender Therapie kontrolliert werden (fakultativ). Im Falle des Auftretens klinischer Zeichen einer Leberfunktionsstörung sind die entsprechenden Kontrollen obligat durchzuführen. Bei einem Anstieg der Lebertransaminasen über das Dreifache der Normwerte sollte Natalizumab ausgesetzt werden. Bei Rückkehr der Transaminasenwerte in den Normbereich kann die Natalizumab-Therapie fortgeführt werden. Die Leberwerte müssen dann engmaschig kontrolliert werden. Bei einem wiederholten Anstieg der Lebertransaminasen über das Fünffache der Normwerte muss Natalizumab permanent abgesetzt werden.
- Alle sechs Monate sollte der JC-Virus (JCV)-AK-Index überprüft werden (obligat), um die individuelle Risikosituation für die Entwicklung einer PML besser abschätzen zu können (siehe Besondere Hinweise).
Radiologische Kontrolle
Zur Beurteilung des Behandlungserfolgs sowie zur möglichen Einschätzung differentialdiagnostisch relevanter Komplikationen der Therapie sollte nach dem Basis-MRT des Schädels mit Kontrastmittel zum Behandlungsbeginn einmal jährlich in den ersten beiden Jahren (d.h. nach zwölf und 24 Monaten) eine MRT des Schädels durchgeführt werden (obligat). Auf die Kontrastmittelgabe sollte hierbei verzichtet werden, wenn es keinen klinischen Anhaltspunkt für einen Krankheitsprogress gibt und wenn eine standardisierte Ausgangs-MRT vorliegt. Sollte die Therapie nach zwei Jahren fortgeführt werden, müssen Maßnahmen höchster klinischer Vigilanz beachtet werden (obligat). Inwieweit engmaschigere MRT-Untersuchungen (z.B. in drei- bis sechsmonatigen Abständen) zur besseren Risikokontrolle beitragen, ist gegenwärtig nicht sicher belegt, dennoch sollte bei Patienten mit entsprechender Risikokonstellation eine solche engmaschigere MRT-Überwachung erwogen werden (Empfehlung des Paul-Ehrlich-Instituts, 03/2016). Häufigere MRT-Untersuchungen können aber für die bessere Vergleichbarkeit sich über die Zeit ändernder zerebraler Befunde hilfreich sein. Idealerweise finden die Verlaufsuntersuchungen am selben Gerät mit denselben Sequenzen statt.
Durchführung und Monitoring während der Gabe
Die Dauer der Infusion beträgt eine Stunde, die Nachbeobachtung eine weitere Stunde. Auch nach der Injektion ist eine Nachbeobachtung für eine Stunde obligat. Maßnahmen und Infrastruktur für die Akuttherapie anaphylaktischer Reaktionen müssen vorgehalten werden (obligat).
Während der Therapie
Schübe, die unter Natalizumab auftreten, können nach Standardvorgaben mit einer Methylprednisolon-Pulstherapie behandelt werden (unter begleitender Weiterführung der Natalizumab-Therapie). Bei atypischer Präsentation sollte differentialdiagnostisch immer an eine PML gedacht werden. Relevant ist hier die Prüfung von Ursachen für ein mögliches zugrunde liegendes Therapieversagen (z.B. das Vorliegen von neutralisierenden Antikörpern gegen Natalizumab). Sollte sich der Verdacht auf eine PML erhärten, muss eine differentialdiagnostische Abklärung mittels MRT und Liquorpunktion (Nachweis von JCV-DNA) unmittelbar angeschlossen werden (obligat).
Die Behandlung Steroid-refraktärer Schübe mittels Apherese-Verfahren sollte nur in erfahrenen Zentren durchgeführt werden, da im Rahmen des Apherese-Verfahrens auch eine Elimination von Natalizumab erfolgt, mit dem Risiko wiederkehrender/fortgesetzter Krankheitsaktivität.
Reevaluation || nach 24 Monaten
- Die Weiterbehandlung nach 24 Monaten kann bei fehlender Kontraindikation erfolgen, wenn die Natalizumab-Wirksamkeit weiterhin belegbar, eine anhaltende Immunkompetenz gegeben und die Indikation weiterhin zu stellen ist (obligat).
- Die Gabe über 24 Monate hinaus ist mit dem Patienten gemeinsam ausführlich zu diskutieren und die Einwilligung in Schriftform zu dokumentieren (obligat; Aufklärungsblatt 24 Monate).
- Die mangelnde Eignung anderer Therapeutika zur Behandlung der aktiven MS sowie die Unwirksamkeit weniger riskanter Therapieoptionen (Therapieversagen unter vorangegangener Therapie für milde / moderate Verlaufsformen der MS) sollten dokumentiert werden (fakultativ).
- Therapiepausen („drug holidays“) stellen keine vernünftige Maßnahme zur Reduktion des Risikoprofils der Therapie bzw. des Risikos opportunistischer Infektionen dar und sind daher nach aktueller Datenlage außerhalb von Studien nicht empfehlenswert.
- Die Therapie darf bei Anzeichen einer beginnenden PML nicht fortgeführt werden (obligat).
Aus Praktikabilitätsgründen werden hier Monate mit Therapiezyklen (= vier Wochen) gleichgesetzt und zur besseren Verständlichkeit mit Monaten gearbeitet.
Absetzen
Untersuchungen an 1.866 Patienten, bei denen Natalizumab wegen der ersten PML-Fälle im Jahr 2005 abgesetzt wurde, haben gezeigt, dass meist die Krankheitsaktivität wieder auftritt wie vor Beginn der Therapie. Einige kleinere Untersuchungen haben gezeigt, dass bei einigen Patienten die Schubaktivität vorübergehend sogar stärker ausgeprägt sein kann als zuvor. Durch den plötzlichen Wegfall der Hemmung durch Natalizumab kommt es zu einem verstärkten und raschen Einstrom von Entzündungszellen ins Nervensystem. Dies kann bereits 4 Wochen nach Absetzen bis hin zu 6 Monaten danach auftreten.
Besondere Hinweise
Schwangerschaft und Stillzeit
- Prinzipiell sollten Frauen im gebärfähigen Alter auf die Durchführung einer wirksamen Empfängnisverhütung aufmerksam gemacht werden (obligat).
- Bei explizitem Schwangerschaftswunsch einer Patientin kann die Therapie bis zum Eintreten der Schwangerschaft unter strenger Nutzen-Risiko-Abwägung fortgeführt werden, da die Krankheitsaktivität erst zwei bis sechs Monate nach Absetzen des Medikaments wieder zunimmt. Zeitlich fällt dies häufig mit dem zweiten/dritten Schwangerschaftstrimenon zusammen, in dem die Krankheitsaktivität wiederum rückläufig ist. Zudem zeigen die bisherigen Schwangerschaften unter Natalizumab keine wesentlichen Auffälligkeiten im Vergleich zur Normalpopulation. Die Betreuung schwangerer Patientinnen unter Therapie mit Natalizumab sollte an erfahrenen Zentren erfolgen.
- In Fällen von hoher Krankheitsaktivität kann, wenn keine alternative Therapie eingeleitet werden kann (weil z.B. schon eine Schwangerschaft eingetreten ist) oder dies nicht gewünscht wird, unter strenger Nutzen-Risiko-Abwägung und klinischer Kontrolle die Therapie mit Natalizumab fortgeführt werden. Die Therapie sollte in diesem Fall durch ein erfahrenes Zentrum erfolgen. Sollte Natalizumab in der Schwangerschaft weitergeführt werden, sollte die letzte Infusion / Injektion vor der 34. Schwangerschaftswoche gegeben werden. Bei Neugeborenen, deren Mütter während der Schwangerschaft gegenüber Natalizumab exponiert waren, wird eine Überwachung der Thrombozytenzahlen, des Hämoglobins und des Hämatokrits empfohlen. Post partum sollte eine Anschlusstherapie erfolgen.
- Das Risiko für Blutbildauffälligkeiten bei den Kindern ist kleiner, wenn man Natalizumab zuletzt um die 30. SSW infundiert, allerdings kommt es dennoch bei 25% der Mütter zu postpartalen Schüben, selbst wenn in den ersten beiden Wochen nach der Geburt wieder mit Natalizumab begonnen wird.
- Natalizumab geht in die Muttermilch über. Es ist allerdings nicht bekannt, ob dies Auswirkungen auf den Säugling hat. Unter Therapie mit Natalizumab darf daher sicherheitshalber nicht gestillt werden.
Daten zu Schwangerschaften unter Natalizumab stammen aus den inzidentellen Schwangerschaften im Rahmen der Zulassungsstudien oder deren Extensionsstudien. Hier war die Fehlgeburtenrate im üblichen Rahmen, es kam zu keinen vermehrten Missbildungen. Als Teil des Risikomanagementplans für Natalizumab wird ein Schwangerschaftsregister geführt. Ein spezifisches Fehlbildungsmuster ergibt sich nach Daten aus dem firmeneigenen und Deutschen Schwangerschaftsregister sowie einer italienischen Publikation derzeit nicht. Die europäische Zulassungsbehörde hat die Ergebnisse des firmeneigenen Registers in die aktuelle Fachinformation aufgenommen und bewertet sie als unproblematisch (keine erhöhte Fehlbildungsrate / kein Muster an Fehlbildungen). Im Deutschen Schwangerschaftsregister zeigt sich bei 350 exponierten Schwangerschaften (doi: 10.1136/jnnp-2023-332804) kein Hinweis auf eine insgesamt erhöhte Fehlbildungsrate, die Kinder wiesen jedoch ein reduziertes Geburtsgewicht auf („small for gestational age“). Ob bei Exposition in der Frühschwangerschaft die frühe Fehlgeburtsrate erhöht ist, kann noch nicht abschließend beurteilt werden, im firmeneigenen Register war sie nicht erhöht. In einer aktuellen italienischen Publikation war sie gegenüber MS Kontrollen erhöht, lag aber mit 17% im natürlich vorkommenden Rahmen.
Impfungen
Die Wirksamkeit von Impfungen kann während und bis zu drei Monate nach Absetzen von Natalizumab eingeschränkt sein. Die Anwendung von attenuierten Lebendimpfstoffen ist unter der Therapie mit Natalizumab kontraindiziert (obligat). Die Wirkung von Impfungen gegen SARS-Cov2 (COVID-19) mit mRNA oder Vektorimpfstoffen ist nach derzeitigen Daten unter Natalizumab nicht wesentlich eingeschränkt.
Detaillierte Empfehlungen zu Impfungen unter MS-Therapien siehe Spezialsituationen.
Seit 2018 ist ein Totimpfstoff gegen VZV (Shingrix®) zur Verhinderung von Herpes-zoster-Infektionen zugelassen. Die STIKO empfiehlt diese Impfung bei allen Personen > 60 Jahren. Bei Personen mit einer Grundkrankheit oder Immunschwäche (wie z.B. durch eine MS-Immuntherapie) empfiehlt die Kommission die Impfung bereits ab einem Alter von 50 Jahren (Indikationsimpfung). Da Zoster bei der Anwendung von Immuntherapien ein häufigeres Problem darstellen kann, kann individuell überlegt werden, den Impfstoff vor Anwendung einer Immuntherapie einzusetzen (off-label).
Infektionen
Bei akuten Infektionen unter Natalizumab sind unverzüglich Maßnahmen zu Diagnostik und Therapie einzuleiten. Sollten sich Zeichen einer Immunkompromittierung zeigen (z.B. Infekthäufung, Aktivierungen latenter Virusinfektionen, opportunistische Infektionen), ist Natalizumab abzusetzen (obligat).
Progressive multifokale Leukenzephalopathie (PML) und Risikoprofil
Die Anwendung von Natalizumab ist mit einem erhöhten Risiko für die Entwicklung einer PML assoziiert. Diese kann tödlich verlaufen (bisher in ungefähr 23% der Fälle) oder zu einer schweren Behinderung führen. Die Prognose einer Natalizumabassoziierten PML hängt auch vom Intervall zwischen Symptombeginn und Diagnose ab. Anhand retrospektiver Analysen durch den Hersteller konnten mehrere Risikofaktoren für die Entwicklung einer PML identifiziert werden:
- (I) Das Risiko einer PML steigt mit zunehmender Behandlungsdauer an, insbesondere wenn die Behandlung über 24 Monate hinaus fortgeführt wird.
- (II) Zusätzlich erhöht sich das Risiko einer PML, wenn vor der Behandlung mit Natalizumab eine potentiell immunsuppressive Vortherapie gegeben wurde. Dieses Risiko scheint unabhängig von der Dauer, der Art und dem Abstand der vorherigen immunsuppressiven Therapie zu sein. Die ursprüngliche Studie summierte unter „nicht- immunsuppressiv“ folgende, seinerzeit verfügbaren Substanzen: Beta-Interferone, Glatirameracetat, Kortikosteroide und intravenöse Immunglobluline.
- (III) Das Vorhandensein von Antikörpern gegen das JC-Virus ist entsprechend neuerer Daten mit einem erhöhten Risiko zur Entwicklung einer PML assoziiert (s.u.).
Entsprechend muss der Patient (und möglichst auch seine Angehörigen) nach zweijähriger Therapie erneut über die Risiken einer Behandlung – insbesondere über das erhöhte Risiko für eine PML – informiert und über erste Anzeichen einer PML und deren Symptome in Kenntnis gesetzt werden (obligat).
Für den Fall, dass der Verdacht auf eine PML besteht, muss die Gabe von Natalizumab so lange ausgesetzt werden, bis eine PML ausgeschlossen werden kann (obligat). Sollte die klinische Untersuchung eine Zuordnung der neurologischen Symptome hinsichtlich eines MS-Schubs bzw. einer PML nicht erlauben, sind weitergehende Untersuchungen einschließlich einer MRT-Untersuchung des Schädels (mit Kontrastmittel) sowie ggf. des Rückenmarks, Liquoruntersuchungen auf DNA des JC-Virus und wiederholte neurologische Kontrolluntersuchungen durchzuführen (obligat). Auch eine negative JC-Virus PCR aus dem Liquor schließt eine PML unter Umständen nicht aus. Bei klinischem Verdacht sind wiederholte Liquoruntersuchungen notwendig und es wird die quantitative Bestimmung der Virus-DNA im Liquor in einem Referenzlabor empfohlen.
PML und IRIS (inflammatorisches Immunrekonstitutionssyndrom)
Ein IRIS tritt bei Patienten, die mit Natalizumab behandelt wurden, häufig nach dem Absetzen der Therapie oder der Elimination von Natalizumab auf. Inwieweit der Plasmaaustausch (PE) das Eintreten eines IRIS fördert, ist unklar. Ein IRIS kann in jedem Fall auch ohne PE auftreten. In der Regel tritt das IRIS innerhalb einiger Tage bis Wochen nach Plasmaaustausch auf. Es kann zu schweren neurologischen Komplikationen führen und ohne entsprechende intensivmedizinische Maßnahmen tödlich verlaufen. Momentan einzige empfohlene Therapie bei einem IRIS sind hochdosierte Steroide, daneben konventionelle intensivmedizinische Maßnahmen zur Kontrolle eines Hirnödems. Eine entsprechende Überwachung, mit geeigneter Behandlung der damit einhergehenden Entzündungsreaktion, sollte konsequent verfolgt werden (obligat).
Rolle von JC-Virus-Antikörpern
In verschiedenen Studien wurde bei Natalizumab-behandelten MS-Patienten untersucht, wie häufig Serum-Antikörper gegen das JC-Virus auftreten. Bei etwas mehr als 50% der Patienten lassen sich Antikörper im Serum nachweisen.
Ein negativer Antikörpertest kann basierend auf der aktuellen Datenlage zumindest ein Hinweis für ein geringeres PML-Risiko sein. Dennoch muss grundsätzlich auch an die Möglichkeit einer Neuinfektion unter Therapie gedacht werden. Darüber hinaus existieren Fallberichte von falsch-negativen Antikörper-Befunden bei älteren (> 65 Jahre) Patienten. Da nur sehr wenige der positiv getesteten Patienten unter Therapie mit Natalizumab im Verlauf tatsächlich eine PML entwickeln, ist der positiv-prädiktive Wert des Tests formal niedrig.
Darüber hinaus korreliert bei Patienten, die positiv auf Anti-JCV-Antikörper getestet und nicht mit Immunsuppressiva vorbehandelt wurden, die Höhe des Anti-JCV-Antikörper-Titers (Index) mit dem Risiko für die Entwicklung einer PML.
Zum gegenwärtigen Zeitpunkt erscheint ein Test bei den folgenden Gruppen sinnvoll:
- zur Therapieentscheidung vor Beginn einer Therapie mit Natalizumab zur Spezifizierung der individuellen Risikosituation.
- bei Patienten mit erhöhtem PML-Risiko unter Natalizumab, also Patienten mit einer vorangegangenen immunsuppressiven Therapie oder einer Behandlungsdauer mit Natalizumab von über zwei Jahren
- bei zuvor negativ getesteten Patienten zur Erfassung einer möglichen Serokonversion (halbjährliche Untersuchung).
Fazit: Ableitend von diesen Erkenntnissen ist die Erhebung einer JCV-Serologie von Patienten mit mehr als zweijähriger Natalizumab-Behandlung sinnvoll. Das über alle Gruppen gemittelte Risiko einer PML-Entwicklung liegt nach derzeitigem Kenntnisstand (03/2024) bei 3,48:1.000 Patienten. Fällt der Antikörpertest positiv aus, sollte die weitere Therapie in Abhängigkeit des Titers evaluiert werden oder ein Absetzen der Therapie erwogen werden. Ein negativer Test bedeutet bei gegenwärtiger Datenlage allerdings keinesfalls, dass Patienten nicht doch eine PML entwickeln können, und entbindet deswegen nicht von den geltenden und empfohlenen Maßnahmen zur Pharmakovigilanz. Bei einem negativen Befund sollte aufgrund der Möglichkeit einer Serokonversion halbjährlich eine Wiederbestimmung erfolgen (siehe Abbildung 1).
Bei Patienten mit positivem Nachweis von JC-Antikörpern ohne vorangegangene immunsuppressive Therapie kann zusätzlich der JCV-Antikörper-Index, ein standardisiertes Maß für die Quantität der vorhandenen Antikörper, zur weiteren Risikoeinschätzung herangezogen werden. Die Testung sollte halbjährlich wiederholt werden, um einen möglichen Titeranstieg zu erfassen. Je höher der Antikörperindex, desto wahrscheinlicher wird die Gefahr einer PML. Möglicherweise ist auch eine Zunahme des Antikörper-Indexwerts bei JCV-positiven Patienten ein Hinweis auf eine PML. Die Daten hierzu sind bislang jedoch nicht ausreichend. Als „nicht-immunsuppressive“ Vortherapien galten in den zugrunde liegenden Studien: (I) Steroide, (II) intravenöse Immunglobuline, (III) Interferon-beta-Präparate, (IV) Glatirameracetat. Für Patienten, die mit anderen Immuntherapien vorbehandelt wurden, besteht streng genommen derzeit keine (vergleichbar) gesicherte Aussagekraft des JCV-Antikörper-Index.
Seit 2024 steht in Zusammenhang mit der Markteinführung des Natalizumab-Biosimilars TYRUKO® ein zweites Testsystem zur Verfügung (ImmunoWELL JCV-IgG Test™). Im Gegensatz zu dem Testsystem, welches durch den Inverkehrbringer von TYSABRI® unterhalten wird (STRATIFY™ JCV DxSelect®), wurden hier abweichende Grenzwerte zur Risikostratifizierung angegeben (s. Tabelle). Die Übereinstimmung des ImmunoWELL-Tests zum STRATIFY-Test wurde in den der EMA vorgelegten Daten mit 90% ermittelt.
Es gibt trotz der in den Zulassungsdaten hohen Übereinstimmung in der Regelversorgung dennoch vereinzelt Berichte über divergente Ergebnisse zwischen den jeweiligen Tests (negativ in Test A, positiv in Test B). Deshalb wird darauf hingewiesen, dass die verwendeten Antikörper-Tests formal an die jeweiligen Präparate gebunden sind und dass der „zugeordnete“ Test für die Risikostratifizierung herangezogen werden sollte. Unabhängige Testverfahren stehen hier bisher nicht zur Verfügung.
| SRATIFY™ JCV DxSelect® | ImmunoWell JCV-IgG Test™ | |
| Geringes Risiko | ≤0,9 | ≤0,8 |
| Moderates Risiko | 0,9-1,5 | 0,8-1,4 |
| Höheres Risiko | ≥1,5 | ≥1,4 |
Basierend auf den initialen Daten wurde vom Paul-Ehrlich-Institut 2016 eine Risikotabelle, die das PML-Risiko in sog. „Behandlungsepochen“ darstellt, veröffentlicht, die 2021 aktualisiert wurde (siehe Abbildung 1).
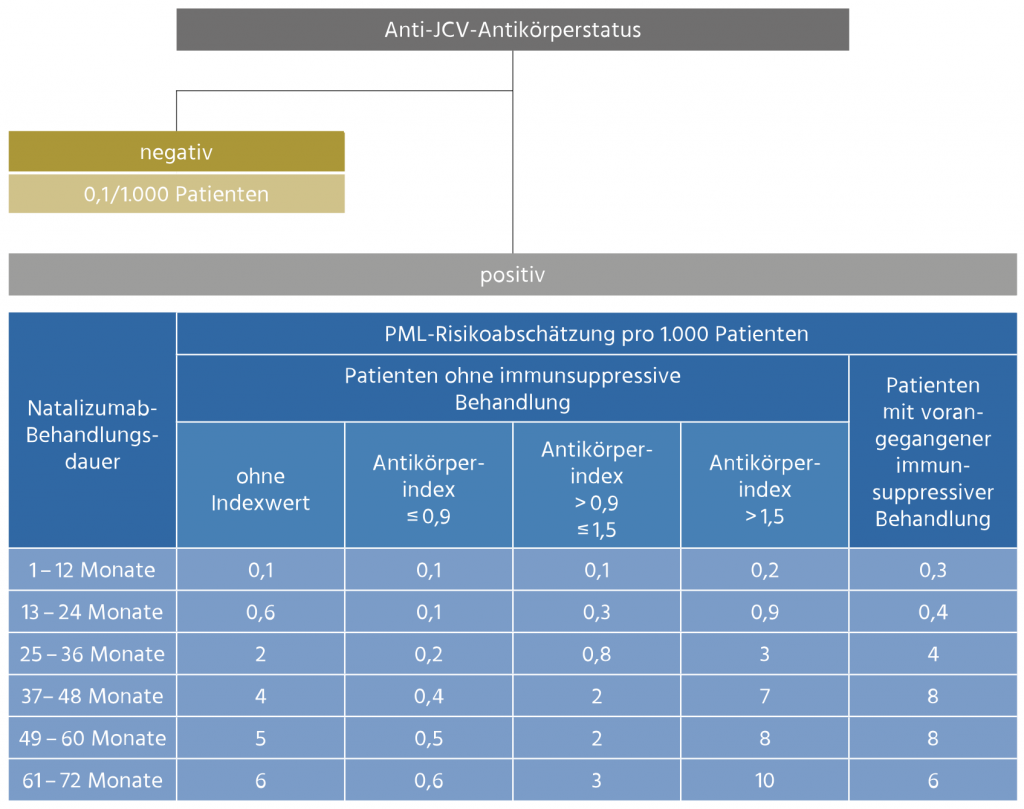
Abbildung 1: PML-Risikoabschätzungen, inklusive Stratifizierung nach Indexgrenzwert bei Anti-JCV-Antikörper-positiven Patienten ohne vorangegangene immunsuppressive Behandlung. Quelle: Arzt-Information & Management-Leitlinien für Patienten mit Multipler Sklerose, die Tysabri® erhalten. Version 22; Genehmigt vom PEI: Januar 2024.
Basierend auf den offiziellen Daten zur PML-Inzidenz (Stand 2017) sowie der Verteilung weiterer Risikomarker wurde eine Projektion des individuellen Risikos veröffentlicht (siehe Abbildung 2). Die dort projizierten Risikokonstellationen wurdenin der Auswertung von 4 großen open-label Studien mit insgesamt 37.249 Patienten weitgehend reproduziert (Ho PR, etal. Lancet Neurol. 2017;16(11):925-933).
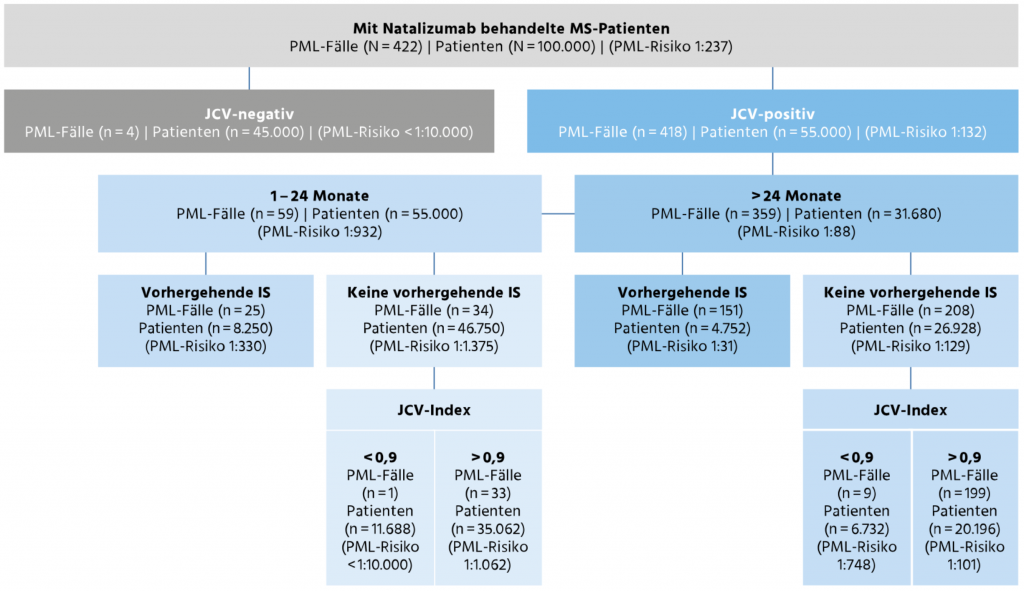
Abbildung 2: Aktualisierte statistische Projektion des individuellen PML- Risikos basierend auf verfügbaren Daten zu Inzidenz sowie relativer Verteilung entsprechender Risikomarker aus dem Zeitraum 2014 – 2016. Diese Daten beziehen sich auf Befunde, die mit dem STRATIFY-Test erhoben wurden und die seinerzeit verfügbaren Daten zur Gesamtinzindenz der Natalizumab-PML. Quelle: Schwab N, et al. Neurology. 2017; 88(12):1197-1205.
In der Vergangenheit wurden verschiedene zusätzliche Labortests zur Einschätzung des individuellen Risikos einer PML im Verlauf der Behandlung evaluiert. Dies schloss sowohl Blut-Tests (CD62L-Dichte auf T-Helferzellen) als auch Untersuchungen des Nervenwassers (Nachweis von sog. „Lipid-spezifischem IgM“) ein. Aktuell steht jedoch keines dieser Verfahren in der Regelversorgung flächendeckend zur Verfügung.
Dauer der Therapie
Für die Behandlung mit Natalizumab existieren sehr robuste Daten zum Nutzen-Risiko-Profil. Die Entscheidung über eine Weiterbehandlung mit Natalizumab nach zwei Jahren sollte in Abhängigkeit von der individuellen Wirksamkeit des Medikaments, unter Berücksichtigung der dann verfügbaren Sicherheitsdaten, möglicher Alternativen und des individuellen Risikos des Patienten erfolgen (siehe auch PML-Risiko unter „Besondere Hinweise“ und „Re-Evaluation nach 24 Monaten“). Der Patient muss darüber hinaus erneut aufgeklärt werden (Patientenaufklärungsbogen zur Langzeittherapie). Eine Weiterbehandlung kann erfolgen, jedoch müssen alle klinischen und paraklinischen Maßnahmen der Pharmakovigilanz beachtet werden.
Autoren
- Prof. Dr. Dr. Sven Meuth
Neurologische Klinik, Heinrich-Heine-Universität Düsseldorf
- Prof. Dr. Heinz Wiendl
Klinik für Neurologie und Neurophysiologie, UNIVERSITÄTSKLINIKUM FREIBURG
- PD Dr. Steffen Pfeuffer
Klinik für Neurologie, Universitätsklinikum Gießen
Weitere Informationen unter „Credits“.
Patientenaufklärungen
Das KKNMS stellt Patientenaufklärungsbögen zur Therapie mit Natalizumab für Sie bereit.
Patientenaufklärung zur Therapie von bis zu zwei Jahren mit Natalizumab (PDF)
Patientenaufklärung zur Langzeittherapie mit Natalizumab (über mehr als 24 Monate) (PDF)
Patientenaufklärung zur Beendigung der Behandlung mit Natalizumab (PDF)
Autoren
Prof. Dr. Dr. Sven Meuth
Neurologische Klinik, Heinrich-Heine-Universität Düsseldorf
Prof. Dr. Heinz Wiendl
Klinik für Neurologie und Neurophysiologie, UNIVERSITÄTSKLINIKUM FREIBURG
PD Dr. Steffen Pfeuffer
Klinik für Neurologie, Universitätsklinikum Gießen

 klicken und
klicken und
Verschiedene Publikationen beschreiben Ablagerungen bzw. Signalveränderungen in speziellen Hirnarealen nach mehrmaligen Kontrastmittelgaben. Ein Krankheitsbild oder Symptome sind auf diese jedoch bislang nicht zurückzuführen. Das KKNMS empfiehlt, bei der Diagnosestellung weiterhin gadoliniumhaltige Kontrastmittel einzusetzen, um die Diagnose nicht zu verzögern und eine aussagekräftige, standardisierte Ausgangs-MRT zu erzielen. Im Krankheitsverlauf kann dann auf die Kontrastmittelgabe verzichtet werden, solange kein klinischer Anhaltspunkt für einen Krankheitsprogress vorliegt und wenn leitliniengerechte MRT-Kontrollen unter Therapie routinemäßig durchgeführt werden.